Research in the Lab
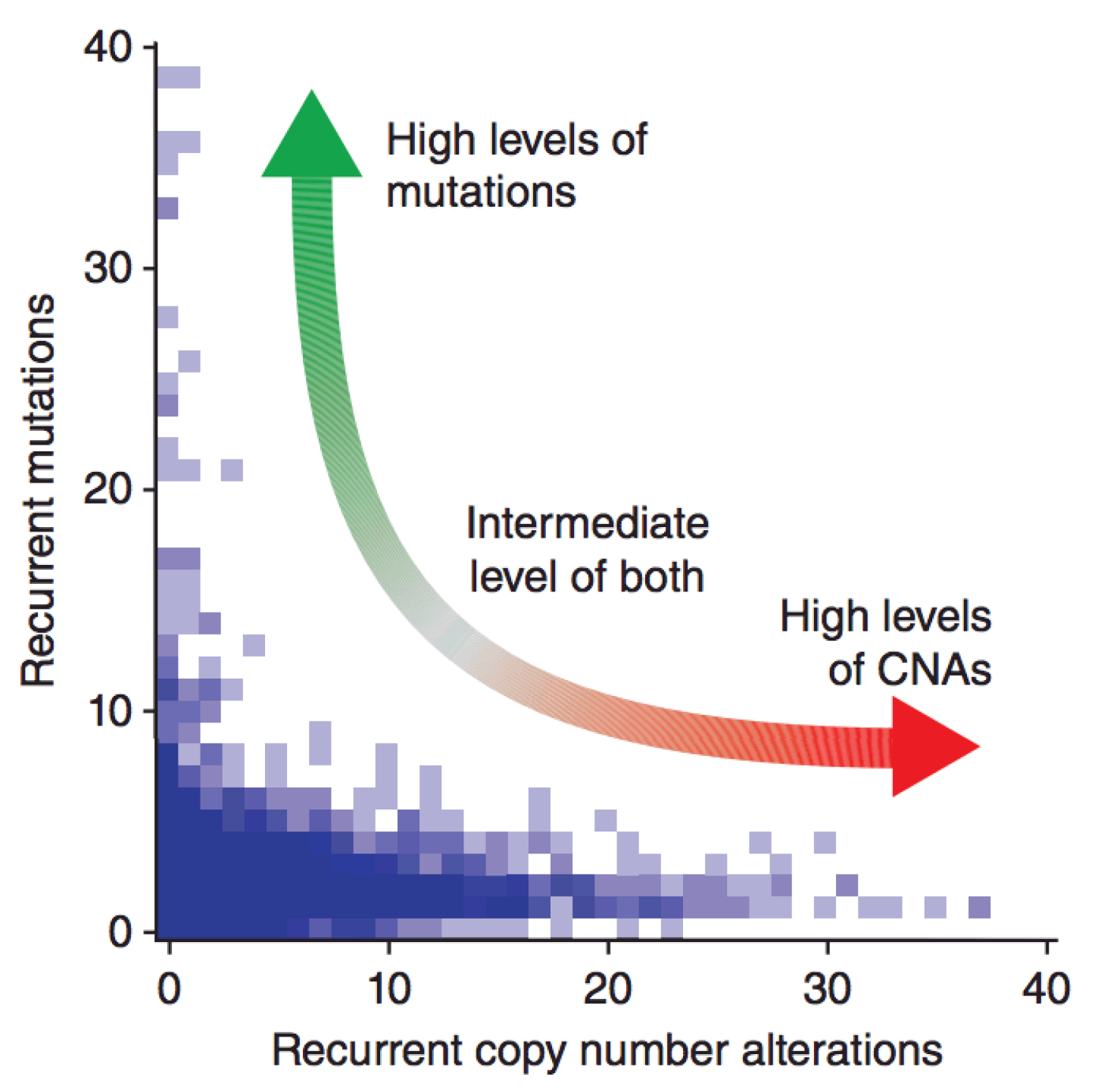
Genomic Dependencies
Cancer emerges through the occurrence and selection of molecular alterations.
A main goal in our group is to understand if and how alterations present in a cancer cell
favor or veto the selection of specific new ones, thus generating non-random patterns of occurrence
across tumor populations.
We observed interdependencies between selected alterations in cancer at multiple levels.
- Distinct mutational processes contribute to tumor insurgence, however extreme genomic instability show a distinctive prevalence of either chromosomal aberrations or somatic mutations.
- Tumor molecular subtypes frequently display characteristic molecular features suggesting the possibility of defining cancer classes based on specific combinations, or signatures, of alterations (Ciriello et al. Nat. Genetics, 2013).
- Finally, protein functional interactions in a cell establish synergisms, redundancies, and incompatibilities between selected alterations. The identification of non-random patterns of occurrence between these events is crucial to highlight functional relationships (Mina et al. Cancer Cell 2017), (Ciriello et al. Genome Res. 2012).
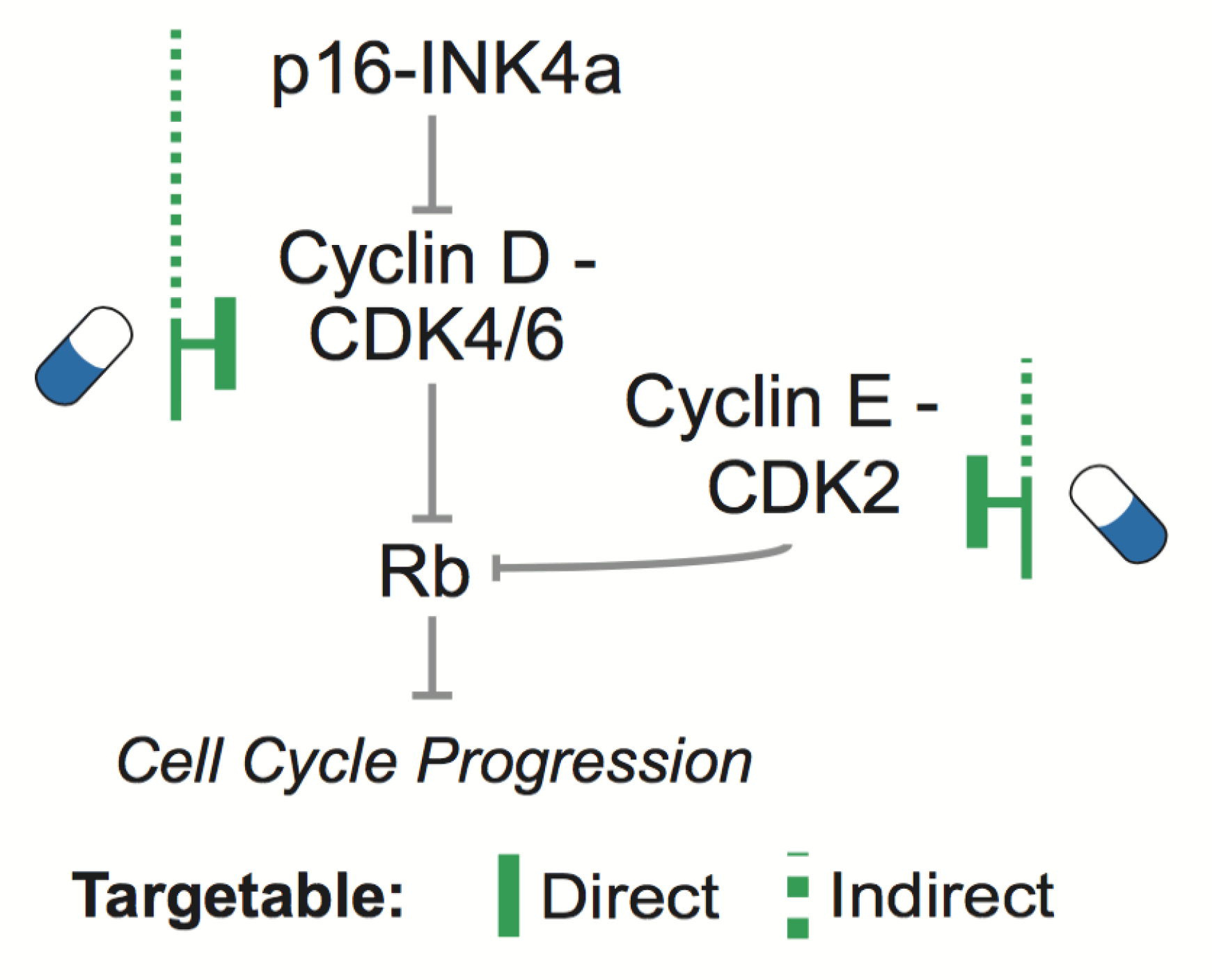
Actionable Cancer Genetics & Epigenetics
Selected genetic and epigenetic modifications in cancer represent implementations of oncogenic mechanisms
and actionable targets for precision treatments.
We dissect large-scale genomic datasets to nominate selected events and identify oncogenic signatures informing the design
of combination therapies.
The complexity of cancer genomes can be distilled into a reduced set of selected alterations with direct biological and clinical interpretation.
Starting from this assumption, we integrate statistical models of recurrence and prior biological knowledge
to identify putative functional genetic and epigenetic modifications.
In particular, we focus on those events that are therapeutically actionable either directly or indirectly, in specific cellular contexts.
Molecular biology studies in this direction focus on breast cancer (Ciriello et al. Cell, 2015),
lymphoma (Oricchio et al. JEM, 2014), and more recently skin melanoma.
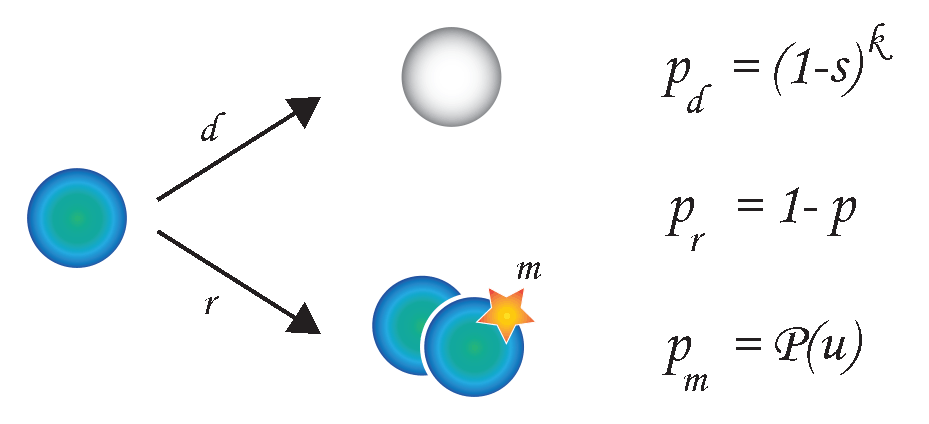
Cancer Evolution
Tumor evolution is typically modeled according to Darwinian principles. By integrating human data with simple models of cancer evolutions,
we aim at exploring which features associate and drive tumor intrinsic heterogeneity and to what extent this can be predicted.
While single-cell genomics and multiple-biopsy studies have the power to provide experimental endpoints to assess tumor heterogeneity,
these are still limited in number and extent. Here, we use prediction of tumor phylogenies from larde-scale genomic profiles
from The Cancer Genome Atlas to estimate which molecular features associate with clonal heterogeneity.
In parallel, we develop numerical models of cancer evolution to monitor the emergence of such heterogeneity as a function
of specific parameters.
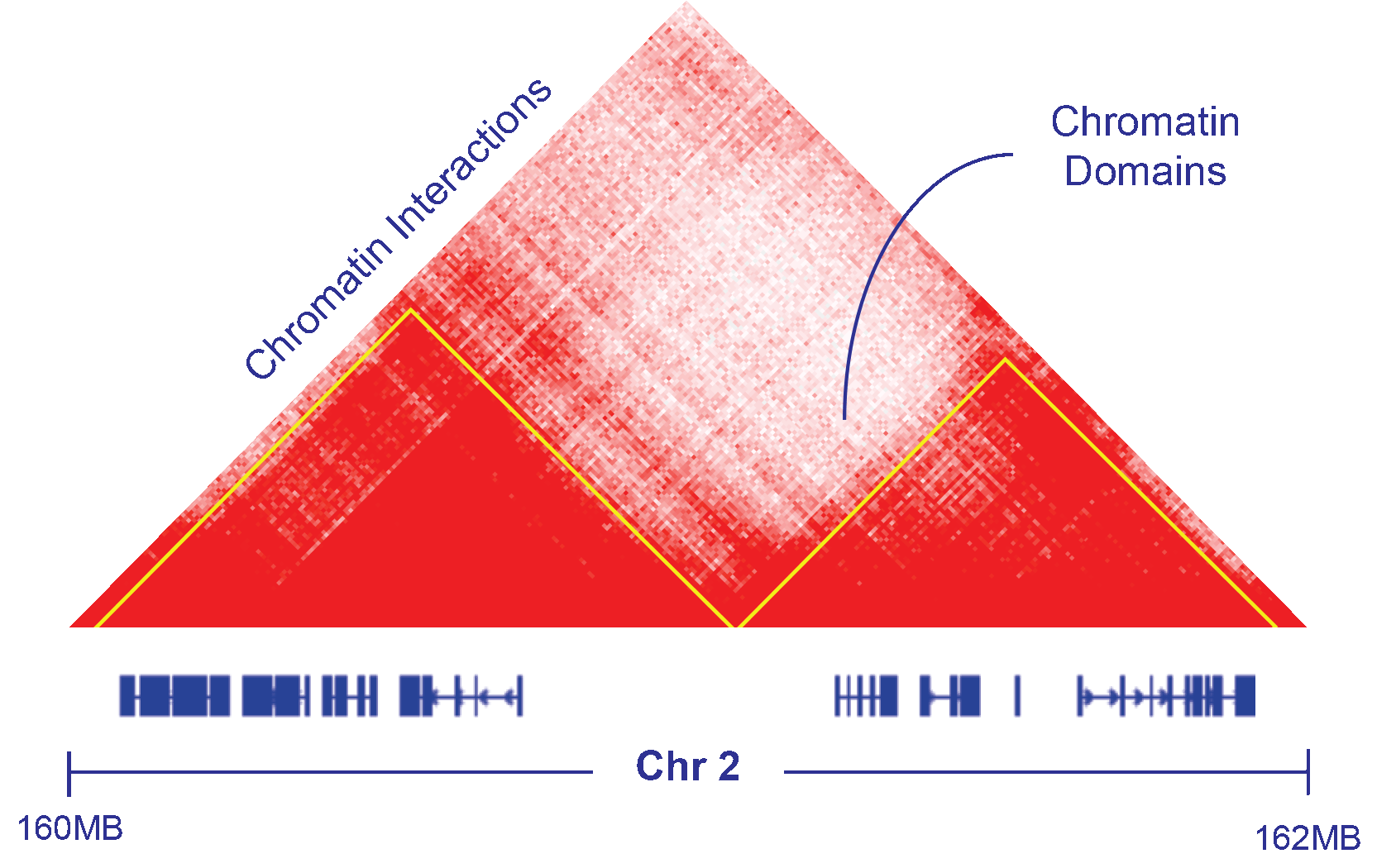
Chromatin Architecture
DNA 3D structures have recently become accessible through Hi-chromatin conformation capture techniques (Hi-C). Our group has recently become interested in this approach to explore how cancer alterations and gene regulation depend and/or affect the chromatin structure.
The Cancer Genome Atlas
Our group has been working within The Cancer Genome Atlas (TCGA) research network since its early phases and it continues to do so now that TCGA is concluding its mission. TCGA research groups are currently bringing together all the samples collected and data generated by the consortium over the last decade with the goal of characterizing altered cellular processes and pathways within and across multiple tumor types. The project, dubbed PanCanAtlas, will explore multi-omics data from ~10K samples based on their distinctive cell of origin, oncogenic processes, and altered pathways. Our group is currently co-leading the PanCanAtlas-Pathway effort together with the groups led by Chris Sander (DFCI), Eli Van Allen (DFCI), and Niki Schultz (MSKCC).